Dr. Erzen Hasani
Pediatër
Spitali Amerikan - Prishtinë
email: ehasani@spitaliamerikan.com
tel: 038 221 661
cel: 045 500 792
Ndër metodat më të shpejta dhe të sigurta të detektimit të këtyre sëmundjeve dhe çrregullimeve të lindura është screeningu neonatal.
Screeningu neonatal konsiston në një panel testesh laboratorike dhe ekzaminimesh klinike që kanë për qëllim zbulimin e prezencës ose jo të disa sëmundjeve të lindura, të cilat nëse nuk diagnostikohen dhe nuk trajtohen drejtë me kohë, mund të japin dëmtime të rënda organike e në raste të rënda edhe të çojnë në vdekje brenda moshës pediatrike.
Programi i screeningut neonatal identifikon tek i porsalinduri çrregullimet:
• Metabolike
• Hormonale
• Hemoglobinopatitë
• Çrregullime të tjera gjenetike siç është Fibroza Cistike
• Çrregullimet e dëgjimit
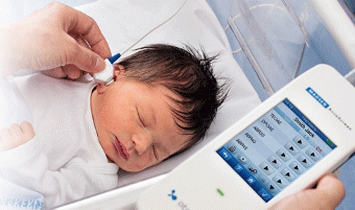
Fatkeqësisht në vendin tonë nuk ekziston një program nacional për screeningun neonatal. I vetmi institucion në Kosovë ku zbatohet ky panel testesh është Spitali Amerikan Kosovë.
Në Spitalin Amerikan në Kosovë, për çdo të porsalindur bëhet screeningu për:
- Çrregullimet e lindura të metabolizmit – ku verifikohen 26 çrregullime të ndryshme të lindura të metabolizmit
- Hipotireozës Kongjenitale – me përcaktimin e hormoneve të gjëndrës tiroide: TSH dhe FT4
- Screeningu i dëgjimit tek i porsalinduri me TeOAE
- Screeningu i icterit (verdhëzës) neonatale në dinamikë – me përcaktimin e bilirubinës totale dhe bilirubinës direkte.
Si bëhet screeningu për çrregullime të lindura të metabolizmit?
Mbas 24-48 orëve të para të jetës, merren disa pika gjak periferik nga thembra e të porsalindurit, pikohen në një letër filtri special – Guthries papper dhe pas tharjes dërgohen në laborator të specializuar. Përpunimi i mostrave bëhet me metodën: Tandem Mass Spectrometry ( MS/MS). Në rast të rezultateve pozitive ose të dyshimta, procedura duhet të përsëritet (Confirmation Test). Rezultati pozitiv nuk do të thotë që fëmija është definitivisht i sëmurë, por që ky fëmijë duhet t’i nënshtrohet testeve specifike diagnostike për patologjinë e caktuar.
Pra, konfirmimi i diagnozës bëhet me marrjen e mostrave të gjakut, urinës e ndonjëherë edhe të lëkurës.
Sipas Komitetit Gjenetik dhe Akademisë Pediatrike Amerikane, screeningu konsiston në 5 faza:
Faza 1: screenimi laboratorik neonatal
Faza 2: ndjekja e rasteve me screenim metabolik laboratorik jonormal dhe menaxhimi i tyre
Faza 3: testimi diagnostik i rasteve pozitive
Faza 4: menaxhimi i sëmundjeve, i cili konsiston në mjekimin terapeutik dhe konsultat gjenetike
Faza 5: evaluimi i vazhdueshëm dhe përmirësimi i sistemit të screenimit neonatal.
Pse është mandator ky screening në vendet e zhvilluara në botë?
- Është metoda më e lirë, më pak agresive dhe traumatike për të porsalindurin dhe familjen.
- Është shumë e shpejtë dhe me saktësi të lartë.
- Mund të performohet në çdo maternitet dhe spital, mjafton të kemi letrën filterin special ku vendosen disa pika gjak dhe më pas të dërgohet më postë në laboratorin adekuat.
- Mundëson diagnostifikimin e hershëm pra diagnoza e hershme do të thotë trajtim terapeutik dhe dietal të hershëm në mënyrë që të minimizohen (eliminohen) pasojat e dëmshme të deficiencës metabolike.
- Diagnoza dhe trajtimi i saktë dhe me kohë, në shumicën e deficiencave garanton një përmirësim të dukshëm të jetës dhe zhvillimit psiko-fizik të pacientëve të prekur. Bile, për disa nga këto deficienca si p.sh. Hipotireoza kongjenitale, e cila nëse diagnostikohet brenda muajit të parë të jetës, trajtohet me hormone të tiroides dhe këto foshnja rriten plotësisht të shëndosha.
Si bëhet screeningu për çrregullimet e lindura të dëgjimit?
Sipas statistikave dëmtimi bilateral i dëgjimit në grupin e të porsalindurve me rrezik normal është 1-3 në 1000 të porsalindur. Në grupin e të porsalindurve me rrezik të lartë, pra të porsalindurit e trajtuar në Njësinë Inten
sive Neonatale, është 2-4 në 1000.
Screeningu bëhet me anë të TEOAE (Transient Evoked Otoacustic Emissions) dhe që në të vërtetë janë përgjigje akustike e veshit të brendshëm ndaj stimulimit akustik të shkurtër me frekuencë të gjerë. Përparësitë e kësaj metode janë se është e shpejtë, joinvazive, sensitive dhe objektive.

- Hipotireoza Kongjenitale (CH): Incidenca 1:4000; Emri i vjetër Kretinizëm, rrjedh nga retardimi mendor dhe fizik i pacientëve të patrajtuar. Shkaku themelor në botë është deficienca e jodit! Në vendet e zhvilluara shkaktar janë kombinimet e faktorëve të njohur dhe panjohur. Më së shumti kemi të bëjmë me defektin në zhvillimin e gjëndrës tireoide atireosa; tiroidea hipoplastike ose tireoidea ektopike. Simptomat: përgjumësia, tonusi muskulor i ulur, zëri i shterur, peristaltika e ngadalësuar, verdhëza e zgjatur, temperaturë e ulët trupore. Tek rastet me atireozë: makroglosi, herni umbilikale, fontanella anterior e madhe. Nëse nuk diagnostikohet dhe trajtohet në muajin e parë të jetës zhvillohet retardim mental me IQ ≤80, ngecje në rritje. Trajtimi me Thyroxine tbl.10-15 µg/kg/ditë. Prognoza për rastet kur trajtimi fillon në javët e para është ekselente.
- Hyperplasia Kongjenitale adrenale (Sindroma adrenogjenitale)(CAH): trashëgohet në mënyrë autosome recesive. Për shkak të mutacionit të trashëguar të gjeneve, gjëndra mbiveshkore nuk prodhon kortisolin nga cholesterol. Hormonet e çrregulluara janë :
- aldosteroni-mineralokortikosteroid
- testosterone-androgjen
- estradioli – estrogjen
Çrregullimet metabolike
Janë pasojë e deficiencës totale apo të pjesshme të enzimeve që marrin pjesë në metabolizimin e substancave të ndryshme në organizëm. Këto substanca marrin pjesë në hallka të ndryshme të zinxhirit metabolik siç janë : aminoacide të ndryshme, acide yndyrore, acide organike, mono apo disaharide. Kjo deficiencë dëmton aftësinë e organizmit për të përdorur substancat e marra me dietë apo të sintetizuara në organizëm. Si rrjedhojë, mund të vijë deri tek akumulimi i këtyre substancave në organizëm dhe për pasojë ka manifestimin klinik të sëmundjes dhe dëmtimin e organizmit.
Në Spitalin Amerikan në Kosovë bëhet skanimi për këto çrregullime të lindura të metabolizmit (Inborn errors of metabolism-IEOM):
Çrregullimet e metabolizmit të amonoacideve:
- Fenilketonuria (PKU) - që është dhe çrregullimi i parë metabolik i identifikuar nga Robert Guthrie qysh në vitin 1960. Trashëgohet në mënyrë autosome recesive. Incidenca:1:10000-15000 SHBA. 1:2600 Turqi.
- Maple syrup urine disease (MSUD) - trashëgohet në mënyrë autosome recesive. Incidenca: 1:18500 ; 1:380 tek Old Order Mennonite. Si pasojë e mutacionit të trashëguar të katër gjeneve vjen deri tek rritja e koncentrimit të aminoacideve Leucine, Isoleucine dhe Valine.
- Tyrosinemia TRY-I, TRY-II, TRY-III – trashëgohet në mënyrë autosome recesive. Incidenca: 1:1846 Quebec-Kanada; 1:100000.
- Homocystinuria (Hypermethionemia) – trashëgohet në mënyrë autosome recesive, Incidenca: 1:6400 Norvegji; 1:17800 Gjermani; 1:1800 Qatar. Si pasojë e deficiencës së cystathionine ß synthase vjen deri tek rritja e homocystinës dhe methioninës. 2 tipe: Tipi I : B6 reaktive, Tipi II: Homocistinuria B6 joreaktive .
- Hyperglycinaemia (Encephalopathia Glycine) NKH – trashëgohet në mënyrë autosome recesive, Incidenca: 1:55000 Finlande, rritja e Glycines aminoacid i nevojshëm për ndërtimin e proteineve, por edhe neurotransmiter. Dallohen 3 forma: Forma klasike, Forma Infantile, Forma atipike.
- Hyperornithinemia (Ornithine Translocase Deficiency With Gyrate Atrophy) HHH – trashëgohet në mënyrë autosome recesive, karakterizohet me humbje progresive të shikimit deri në moshën 50 vjeçare.
Çrregullimet e Ciklit të Uresë:
- Citrulinemia (Arginosuccinate synthetase deficiency) - trashëgohet në mënyrë autosome recesive. Tipi 1 –klasik vërehet që në ditët e para të jetës dhe karakterizohet me letargji, vështirësi në thithje, vjellje, konvulzione. Tipi 2 –afekton SNQ, karakterizohet me konfuzion, iritabilitet, konvulzione deri në komë, (Encephalopati hepatike). Mund të inicohet edhe nga disa barna, infeksionet, alkoholi. Zhvillohet edhe tek personat të cilët kanë patur cholestaze nenatale. Trajtimi: dietë me pak proteinë dhe Sodium Phenylacetate I.V.
- Arginino Succinik Aciduria (ASL Deficiency)(ASA) - trashëgohet në mënyrë autosome recesive. Simptomat janë letargji, vjellje, hipotermi, hiperventilim, hepatomegali dhe encefalopati progresive. Trajtimi: dietë me pak proteinë.
- Arginemia (Arginase Deficiency) - incidenca 1:300000, trashëgohet në mënyrë autosome recesive. Simptomat paraqiten zakonisht në moshën 3 vjeçare: spasticitet i ekstremiteteve, humbje e kontrollit të zorrëve dhe fshikëzës urinare, retardimi mental i rëndë, konvulzione. Trajtimi: Akut: dializa; sodium Phentlacetate, Sodium Benzoate; dietë e pasur me karbohidrate dhe yndyra.
/Telegrafi/